

Saving MDMA VII: This isn't The First Drug to Have Problems Getting Approved
Saving MDMA VII: This isn't The First Drug to Have Problems Getting Approved
A bedtime story about another first-in-class drug.

The Frontier Psychiatrists is a daily enough health-themed newsletter. Recently, I've done quite a bit of writing around the FDA approval process for the Lykos Theraputics Breakthrough Submission of MDMA-AT. That stands for MDMA-assisted therapy. The pretense is that this breakthrough treatment is a combination of an old off-patent drug, MDMA, and manualized Psychotherapy. I've already written about how I think the therapeutic component of this is not good. Like others, I have written about some of the very serious problems in the clinical trials from both an ethics and an FDA perspective. Here are those prior articles—subscribe! Some are behind a paywall now—you snooze, am I right?
The “Saving MDMA” Series!
To put my dime down, I don't think MDMA-AT will be approved by the FDA when it meets to discuss the 10-1 and 9-2 against AdComm votes cautioning against approval of the combination product at this time. For most well-capitalized pharma companies, this is part of the process.
The most crucial reason this will happen? The FDA will find it expedient to inform the study sponsor that they don't have the safety data necessary to approve the drug. They can then kick the can down the road. They can buy time to finish their investigation into alleged misconduct in the trials. This is not a hard call for the FDA. Lykos was asked to provide safety data — EKG and laboratory work — and they didn't. So, the FDA is well within its rights to say go back and get that for us, at a minimum. Remember, the FDA is staffed by humans. Those humans have a job to do, to make sure the boxes are checked when it comes to the safety of any new therapeutic. They also have every other new drug and Device to review and issue an opinion on, and they can't spend all day on MDMA.
Today's column is using the above as a pretext, but it's telling the story of another drug. I understand for many enthusiasts of psychedelic medicines, this is their very first FDA approval process. I remember my first time. I think we all do. The first time, we flipped through accessfda.gov, reviewing the submitted data and looking— longingly— at the extremely guarded and careful language the FDA uses to communicate with study sponsors of clinical trials. First times are memorable, but for some of us, like your author, this is not the first therapeutic we have watched, from afar, go through a complex regulatory process.
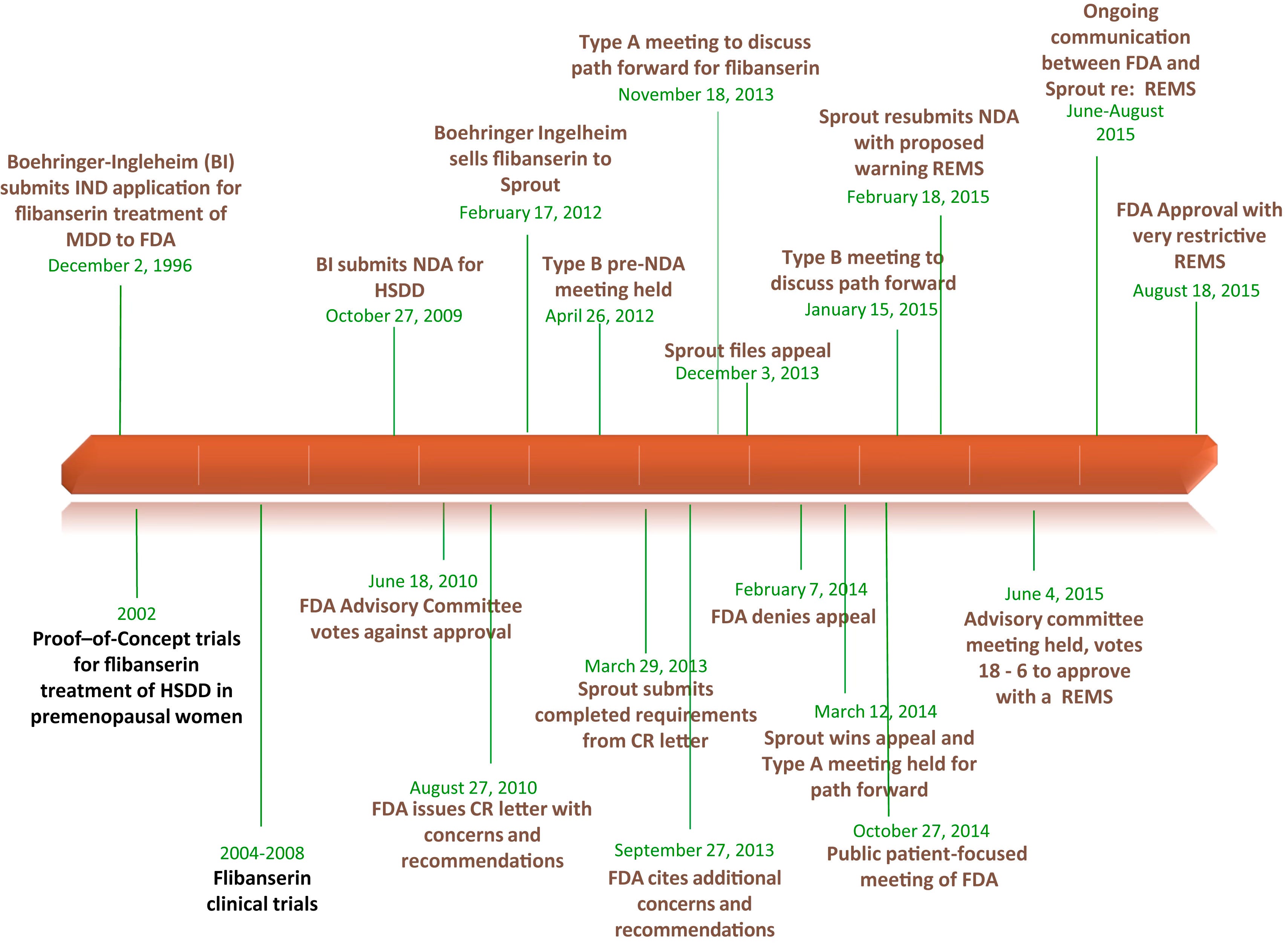
Today, I'm going to tell the story of a drug called Flibanserin (Brand Name: Addyi). This, too, was a novel therapeutic. It's a drug that was supposed to address a brand new disorder—with no other treatments! What happened in the course of this drug’s approval process is instructive. The FDA had a new disorder to evaluate a new treatment for, and to ruin the punchline; it took a while.
Hypoactive Sexual Desire Disorder made its debut in DSM-IV-TR:
Hypoactive Sexual Desire Disorder (HSDD) is defined in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Revised (DSM-IV-TR) as persistent deficient sexual fantasies and desire for sexual activity that causes marked distress or interpersonal difficulty.1
There were lots of drugs for male sexual dysfunction already. Viagra, of course, was the star. And the political pressure was on the FDA was to not leave women behind. An upstart pharmaceutical company, Sprout Pharmaceuticals, had a novel compound that they believed was a solution to this problem for women. A female Viagra. Finally, gender equality is in pill form!
From a mechanism standpoint—it’s not remotely Viagra. That is a drug that improves blood flow to the penis in people who want to have sex but can’t. On the other hand:
flibanserin is a non-hormonal, centrally acting daily medication that changes the balance of the neurotransmitters that affect sexual desire2
It's worth noting, for readers unfamiliar with the data on Viagra (sildenafil), it's not such a good drug either. Viagra, and so many other drugs, was a happy accident. It was initially developed as a blood pressure medication, and then it was noticed that increased erectile function was a common adverse effect. However, many men don't refill the Viagra prescription after the first script, and far less than half are taking it three years later:
The response rate for sildenafil treatment was 67%. Of these sildenafil responders, 43% reported that they continued using sildenafil while 57% did not, in a mean follow-up of 3 years. Common reasons for discontinuation were effect below expectations, high cost, loss of interest in sex, and inconvenience in obtaining sildenafil.3
It is not a wildly effective treatment for erectile dysfunction. It is, however, better than nothing. But that never stopped a good drug development processs with a politically expedient market. The playbook was true to pharma form—readers will remember Abbott Labs’ strategy with Depakote for the newly rebranded Bipolar Disorder—create a screen tool (the MDQ) for the problem we intend to medicate!
Using the MDQ as a screening tool results in catastrophic over-identification—only 18% of people who test positive on the MDQ questionnaire would have bipolar disorder at all. Across America, 25.84 million people would screen positive on the MDQ. Of those, only 4.65 million would have manic-depressive illness.4
Sprout had a candidate molecule and a brand new disorder to raise awareness about. They needed a screening tool5! And lo, it came to pass… in the form of the Decreased Sexual Desire Screener (DSDS):

This is not as bad a psychometric as the MDQ:
the DSDS's sensitivity and specificity were 83.6% and 87.8%, respectively.
To make it a little easier for lay audiences:
The false negative rate (FNR) is 16.4%.
The false positive rate (FPR) is 12.2%.
A new disorder, a new screening tool, a new drug—for women! This should be easy!! Unfortunately, it wasn't.
Studies need primary endpoints—what are we measuring to determine if the drug works? This “primary endpoint” is selected in advance of the trial being run, so you can’t just pick one of the 20 endpoints that happened to show benefit after the fact. Functional Sexual Desire (FSD) disorders like Erectile Dysfunction in men had already had some questionable choices made regarding their endpoints—they chose a broad, patient self-reported scale as a primary endpoint for Vigara:
The draft guidance for studies of FSD released by the FDA in 2000 (and withdrawn in 2010) did not consider such broad patient-reported outcomes acceptable efficacy measures in studies of FSD.
For flibanserin, they wanted it documented the sex that was good…which I think we can all agree is one heck of a better standard in science:
the guidance required separate studies or analyses for pre- and postmenopausal women with FSD and required satisfying sexual events (SSEs) as a primary outcome measurement despite objections by sexual medicine experts.
SSEs have the benefit of being a scalar variable—there was one, or two, or three, or—heavens—four. There is a difference between one and two, and two is twice as many as one. It’s not as subjective, in theory, as “50% better sexual events.” It’s not a measurement of how much better you were willing to tell a research assistant you felt about the quality of the experience. It happened, or it didn't—with the caveat of the “satisfying” word.
This—increase in SSEs— was chosen as the primary endpoint for the new drug. There were some problems, however:
An alcohol challenge test result that showed significant problems with hypotension and dizziness with “substantial amounts of alcohol (≥5 units).6”
Yes, a “let’s have more sex drug” also has an interaction—with adverse events—with alcohol. I can’t see any problem there. Neither did the at-the-time owner of the molecule, Pharm Giant (and My Connecticut Hometown Favorite) Boehringer Ingelheim (BI), when they submitted a New Drug Application to the FDA in 2009 for this indication. This is after their initial submission for its possible use as an antidepressant in 1996…but the first AdComm meeting for BI’s submission voted against its approval, and BI offloaded the molecule to Sprout Pharmaceuticals in 2012.
The fight continued for this sort of revolutionary treatment for a novel disorder under Sprout’s stewardship. What followed were more clinical trials, another FDA denial, a proposed Risk Evaluation and Mitigation Strategy Program (REMS), another AdComm Meeting, and eventual approval—with a mandatory REMS—in 2015.
One of the core issues—similar to critiques of the FDA process in the MDMA matter—was the unfamiliarity of the FDA sub-group assigned to evaluate this drug. Recall that Flibanserin was a repurposed antidepressant that works in the brain to restore sexual desire. It was reviewed not by the CNS drug experts but by the FDA Division of Bone, Reproductive, and Urologic Products (DBRUP). They were very concerned about the adverse effects related to:
The incidence of somnolence was 17% with flibanserin 50 mg twice daily, 11% with flibanserin 100 mg at bedtime, and 3% with placebo.7, 13 FDA concerns initially centered on the incidence of sedation (and whether these effects would continue the day after bedtime dosing, causing cognitive impairment and/or sedation-related accidents), syncope, hypotension, and the interaction of flibanserin with alcohol and moderate or strong CYP3A4 inhibitors.7
The drug was eventually approved with a REMS. It also included a “no-alcohol” black box warning. This was later revised by the FDA based on post-marketing data:
the FDA has determined that changes must be made to Addyi’s labeling to clarify that there is still a concern about consuming alcohol close in time to taking Addyi but that it does not have to be avoided completely. Specifically, the boxed warning, contraindication, warnings and precautions, and adverse reactions sections of labeling are being updated to reflect that women should discontinue drinking alcohol at least two hours before taking Addyi at bedtime or to skip the Addyi dose that evening. Women should not consume alcohol at least until the morning after taking Addyi at bedtime.
Which made it a heck of a lot easier to prescribe, I’d imagine?
The cautionary tale and hopeful message for MDMA and other related compounds? The FDA is concerned with safety data. That's their job. They get to regulate the marketing of drugs for approved indications with known safety and adverse event profiles. As long as the study sponsor comport itself appropriately, even drugs with vast array of interactions and adverse effects can be considered for approval. But the FDA has to think about what drugs are gonna be like in the real world, when they're combined with other drugs, with which they may have adverse effects. It's not just what happened in the studies, although that matters, it's what is going to happen at vast scale that they're concerned with.
It's not about one advisory committee meeting, one clinical trial, or one decision. This is a relationship. The FDA has a relationship with study sponsors, and that relationship unfolds over time. It's not a friendship, and it's not adversarial, per se. Most of us don't have regulatory relationships in our lives, so it's hard to imagine what this is like for most people. And as long as you're willing to put in the time and effort—and, in some cases, phenomenal amounts of money—getting a drug that is effective over the line is possible. But you're not guaranteed that approval at that first meeting. Sometimes, it's gonna take years. Yes, people may be suffering in the meantime. That is just the way it goes. Nothing before its time might as well be the motto of the FDA, particularly with novel treatments. The breakthrough pathway was supposed to address some of this delay, and it has for other novel therapeutics. Breakthrough is based on the need—life-threatening conditions—and the ability of the proposed intervention to create better outcomes than the standard of care. That accelerates the process, and improves back-and-forth with the agency, but it doesn't change the fundamental standard. FDA approved treatments should be safe and effective.
The median cost of getting a new drug to maket was $1.3b in a 2020 study.8. Lykos has spent only a tiny fraction of this amount. Either be prepared to spend much, much much more, because that's on average what it costs, or it may be that you don't have the access to capital necessary to bring a new therapeutic to market in the United States?
Can MDMA get over the line? Based on the experience of Flibanserin, of course. There are questions as to whether the juice is worth the squeeze, financially, but hey, this is a revolution, and it’s not about money, it’s about changing human consciousness or something. By the way, does anyone have a spare ticket to the burn?

—Owen Scott Muir, M.D., DFAACAP.
Dooley, E. M., Miller, M. K., & Clayton, A. H. (2017). Flibanserin: From Bench to Bedside. Sexual Medicine Reviews, 5(4), 461-469. https://doi.org/10.1016/j.sxmr.2017.06.003
Laumann, E. O., Paik, A., & Rosen, R. C. (1999). Sexual dysfunction in the United States: prevalence and predictors. Jama, 281(6), 537-544.
Jiann, B., Yu, C., Su, C., & Tsai, J. (2006). Compliance of sildenafil treatment for erectile dysfunction and factors affecting it. International Journal of Impotence Research, 18(2), 146-149. https://doi.org/10.1038/sj.ijir.3901379
Hirschfeld RM. The Mood Disorder Questionnaire: a simple, patient-rated screening instrument for bipolar disorder. Prim Care Companion J Clin Psychiatry 2002; 4:9–11
Clayton, A. H., Goldfischer, E. R., Goldstein, I., DeRogatis, L., Lewis-D’Agostino, D. J., & Pyke, R. (2009). Validation of the decreased sexual desire screener (DSDS): a brief diagnostic instrument for generalized acquired female hypoactive sexual desire disorder (HSDD). The journal of sexual medicine, 6(3), 730-738.
Pfizer Laboratories, Viagra prescribing information 2015, Available at:
http://labeling.pfizer.com/ShowLabeling.aspx?id=652, Accessed March 15, 2017.
Wouters OJ, McKee M, Luyten J (March 2020). "Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018". JAMA. 323 (9): 844–853. doi:10.1001/jama.2020.1166. PMC 7054832. PMID 32125404.
I spent 20 years in pharma in medical affairs and clinical development, and this use case example raises so many important issues in the current regulatory pathway to drug approval. If nothing else FDA should turn it's focus to real world evidence/data instead of it's reliance on current traditional clinical trials.